What Is Pulmonary Arterial Hypertension (PAH)?
Pulmonary Arterial Hypertension (PAH) is a rare but serious cardiovascular disorder characterized by abnormally high blood pressure within the pulmonary arteries. These arteries carry blood from the heart to the lungs for oxygenation. Over time, increased pressure causes the right side of the heart to work harder, potentially leading to right ventricular failure and reduced survival if left untreated.
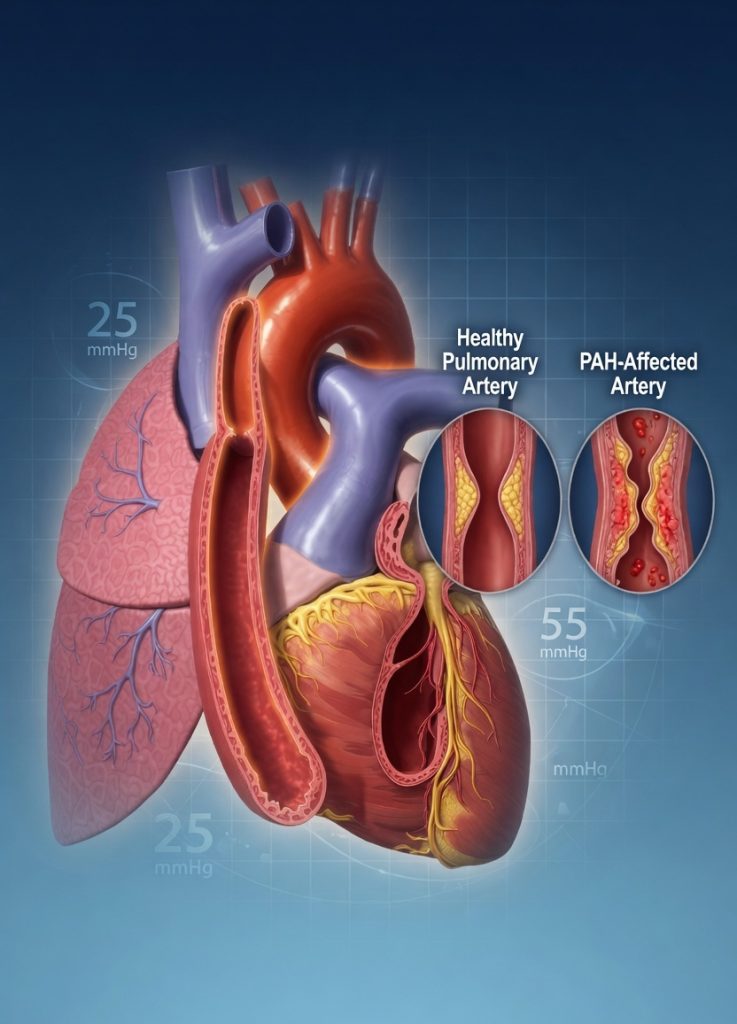
PAH is classified as World Health Organization (WHO) Group 1 Pulmonary Hypertension, distinguishing it from pulmonary hypertension caused by left-sided heart disease, chronic lung disorders, or chronic thromboembolic disease.
Hemodynamic Definition
According to current international guidelines, PAH is defined by:
- Mean Pulmonary Arterial Pressure (mPAP) ≥ 20 mmHg
- Pulmonary Vascular Resistance (PVR) > 2 Wood Units
- Pulmonary Artery Wedge Pressure (PAWP) ≤ 15 mmHg
Accurate diagnosis requires comprehensive clinical assessment and confirmation through right heart catheterization.
Pathophysiology of PAH
PAH develops as a result of progressive remodeling of the pulmonary vasculature. Multiple biological pathways become disrupted, leading to narrowing and stiffening of pulmonary arteries.
Key abnormalities include:
Increased Endothelin-1 Activity
Endothelin-1 is a potent vasoconstrictor that promotes blood vessel narrowing and smooth muscle proliferation.
Reduced Nitric Oxide Production
Nitric oxide normally relaxes blood vessels and inhibits abnormal cell growth. Reduced nitric oxide activity contributes to increased vascular resistance.
Impaired Prostacyclin Pathways
Prostacyclin helps maintain vascular dilation and inhibits platelet aggregation. Deficiency promotes vasoconstriction and vascular remodeling.
Consequences of These Changes
Narrowing of pulmonary arteries
Increased pulmonary vascular resistance
Reduced cardiac output
Progressive right ventricular strain
Right heart failure in advanced disease
Causes and Risk Factors of PAH
PAH may occur without an identifiable cause or develop secondary to other medical conditions.
Idiopathic PAH
No clear underlying cause can be identified.
Associated PAH
PAH may occur in association with:
Connective tissue diseases (systemic sclerosis, lupus)
Congenital heart disease
Portal hypertension and chronic liver disease
HIV infection
Heritable genetic mutations
Exposure to certain medications and toxins
Early recognition of risk factors is essential for timely diagnosis and treatment.
How is PAH Diagnosed?
A comprehensive diagnostic approach is required to confirm PAH and identify potential underlying causes.
Physical Examination
Healthcare providers assess:
- Heart sounds
- Signs of right heart strain
- Peripheral edema
- Oxygen saturation
Echocardiography
Echocardiography is typically the first non-invasive screening tool used to evaluate cardiac structure and estimate pulmonary artery pressures.
Right Heart Catheterization
Right heart catheterization remains the gold standard diagnostic test and is necessary to confirm PAH.
Additional Diagnostic Tests
- Pulmonary function testing
- Six-minute walk test
- CT imaging
- Cardiac MRI
- Blood tests for connective tissue diseases
- Biomarker assessment (BNP or NT-proBNP)
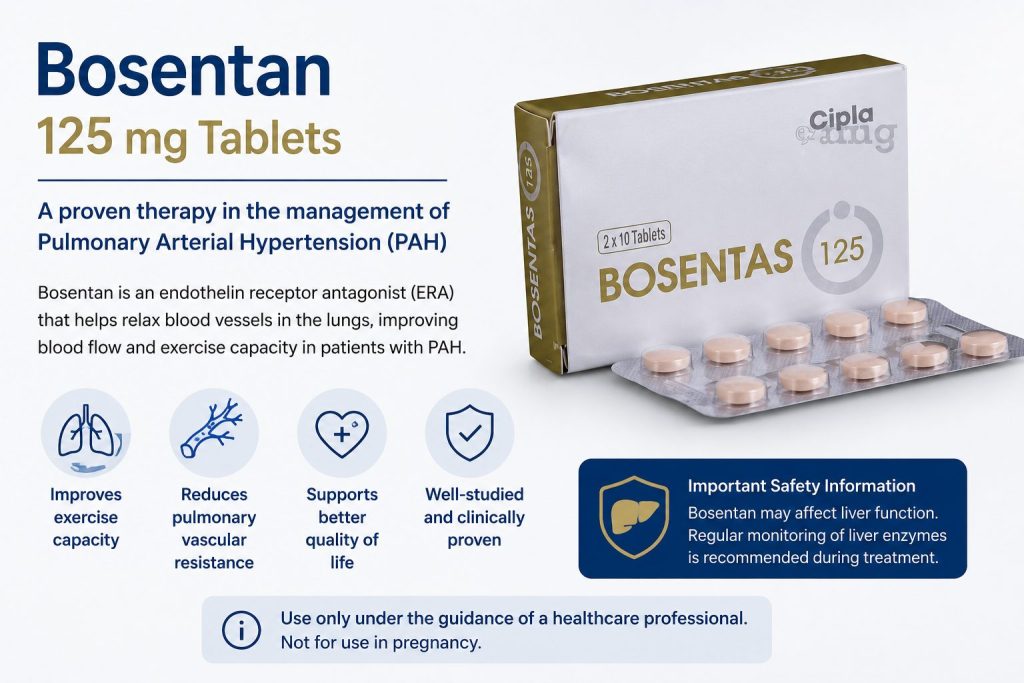
Role of Bosentan in PAH Management
Bosentan is a medication classified as an endothelin receptor antagonist (ERA). Endothelin is a substance in the body that causes blood vessels to constrict and promotes cell proliferation, contributing to the narrowing of pulmonary arteries in PAH.
Bosentan works by blocking the effects of endothelin, helping to relax and widen the blood vessels in the lungs. This reduces pulmonary artery pressure and improves blood flow, which can alleviate symptoms, improve exercise capacity, and slow disease progression.

Benefits of Bosentan
- Improves exercise tolerance and quality of life
- Reduces symptoms such as breathlessness and fatigue
- May delay worsening of PAH when used as part of a comprehensive treatment plan
Important Considerations
Bosentan requires regular monitoring due to potential side effects, including liver toxicity and anemia. Patients on bosentan typically undergo periodic liver function tests and blood counts to ensure safety.
Current Treatment Strategies for PAH (2026 Update)
Modern PAH management increasingly focuses on early intervention and combination therapy.
Emerging strategies include:
- Early combination treatment approaches
- Risk-based treatment selection
- Personalized medicine programs
- Novel endothelin pathway modulators
- Advanced prostacyclin therapies
- Precision monitoring using biomarkers and imaging
These advances continue to improve long-term outcomes for patients with PAH.
Frequently Asked Questions (FAQ)
Is Pulmonary Arterial Hypertension Curable?
Currently, PAH is not considered curable. However, modern therapies can significantly improve symptoms, quality of life, and long-term outcomes.
How Effective Is Bosentan?
Bosentan has demonstrated significant improvements in exercise capacity, symptom control, and delayed disease progression in many patients with PAH.
Can Bosentan Be Used During Pregnancy?
No. Bosentan is contraindicated during pregnancy because it can cause serious birth defects.
How Often Should Liver Function Tests Be Performed?
Healthcare providers typically recommend regular liver function monitoring throughout treatment, particularly during the first months of therapy.
Pulmonary arterial hypertension is a challenging condition, but advances in treatment, including the use of bosentan, have significantly improved patient outcomes. If you or a loved one is diagnosed with PAH, working closely with a healthcare provider specializing in pulmonary hypertension is essential to tailor the best treatment plan.
PAH Treatment Articles
- Riociguat for Pulmonary Hypertension
- Ambrisentan: Uses and Benefits in PAH
- Macitentan Therapy for Pulmonary Arterial Hypertension
- Selexipag for Advanced PAH
Conclusion
Pulmonary Arterial Hypertension is a complex and progressive disease requiring specialized diagnosis and long-term management. Advances in understanding the disease process have led to targeted therapies such as Bosentan, which address key pathways involved in pulmonary vascular remodeling.
Through early diagnosis, evidence-based treatment strategies, regular monitoring, and multidisciplinary care, many patients with PAH can achieve improved symptom control and better quality of life. Patients should work closely with healthcare professionals experienced in pulmonary hypertension to develop an individualized treatment plan.